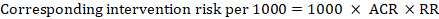
Holger J Schünemann, Julian PT Higgins, Gunn E Vist, Paul Glasziou, Elie A Akl, Nicole Skoetz, Gordon H Guyatt; on behalf of the Cochrane GRADEing Methods Group (formerly Applicability and Recommendations Methods Group) and the Cochrane Statistical Methods Group
Cite this chapter as: Schünemann HJ, Higgins JPT, Vist GE, Glasziou P, Akl EA, Skoetz N, Guyatt GH. Chapter 14: Completing ‘Summary of findings’ tables and grading the certainty of the evidence [last updated August 2023]. In: Higgins JPT, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, Welch VA (editors). Cochrane Handbook for Systematic Reviews of Interventions version 6.5. Cochrane, 2024. Available from www.training.cochrane.org/handbook.
‘Summary of findings’ tables present the main findings of a review in a transparent, structured and simple tabular format. In particular, they provide key information concerning the certainty or quality of evidence (i.e. the confidence or certainty in the range of an effect estimate or an association), the magnitude of effect of the interventions examined, and the sum of available data on the main outcomes. Cochrane Reviews should incorporate ‘Summary of findings’ tables during planning and publication, and should have at least one key ‘Summary of findings’ table representing the most important comparisons. Some reviews may include more than one ‘Summary of findings’ table, for example if the review addresses more than one major comparison, or includes substantially different populations that require separate tables (e.g. because the effects differ or it is important to show results separately). In the Cochrane Database of Systematic Reviews (CDSR), all ‘Summary of findings’ tables for a review appear at the beginning, before the Background section.
Planning for the ‘Summary of findings’ table starts early in the systematic review, with the selection of the outcomes to be included in: (i) the review; and (ii) the ‘Summary of findings’ table. This is a crucial step, and one that review authors need to address carefully.
To ensure production of optimally useful information, Cochrane Reviews begin by developing a review question and by listing all main outcomes that are important to patients and other decision makers (see Chapter 2 and Chapter 3). The GRADE approach to assessing the certainty of the evidence (see Section 14.2) defines and operationalizes a rating process that helps separate outcomes into those that are critical, important or not important for decision making. Consultation and feedback on the review protocol, including from consumers and other decision makers, can enhance this process.
Critical outcomes are likely to include clearly important endpoints; typical examples include mortality and major morbidity (such as strokes and myocardial infarction). However, they may also represent frequent minor and rare major side effects, symptoms, quality of life, burdens associated with treatment, and resource issues (costs). Burdens represent the impact of healthcare workload on patient function and well-being, and include the demands of adhering to an intervention that patients or caregivers (e.g. family) may dislike, such as having to undergo more frequent tests, or the restrictions on lifestyle that certain interventions require (Spencer-Bonilla et al 2017).
Frequently, when formulating questions that include all patient-important outcomes for decision making, review authors will confront reports of studies that have not included all these outcomes. This is particularly true for adverse outcomes. For instance, randomized trials might contribute evidence on intended effects, and on frequent, relatively minor side effects, but not report on rare adverse outcomes such as suicide attempts. Chapter 19 discusses strategies for addressing adverse effects. To obtain data for all important outcomes it may be necessary to examine the results of non-randomized studies (see Chapter 24). Cochrane, in collaboration with others, has developed guidance for review authors to support their decision about when to look for and include non-randomized studies (Schünemann et al 2013).
If a review includes only randomized trials, these trials may not address all important outcomes and it may therefore not be possible to address these outcomes within the constraints of the review. Review authors should acknowledge these limitations and make them transparent to readers. Review authors are encouraged to include non-randomized studies to examine rare or long-term adverse effects that may not adequately be studied in randomized trials. This raises the possibility that harm outcomes may come from studies in which participants differ from those in studies used in the analysis of benefit. Review authors will then need to consider how much such differences are likely to impact on the findings, and this will influence the certainty of evidence because of concerns about indirectness related to the population (see Section 14.2.2).
Non-randomized studies can provide important information not only when randomized trials do not report on an outcome or randomized trials suffer from indirectness, but also when the evidence from randomized trials is rated as very low and non-randomized studies provide evidence of higher certainty. Further discussion of these issues appears also in Chapter 24.
Several alternative standard versions of ‘Summary of findings’ tables have been developed to ensure consistency and ease of use across reviews, inclusion of the most important information needed by decision makers, and optimal presentation (see examples at Figures 14.1.a and 14.1.b). These formats are supported by research that focused on improved understanding of the information they intend to convey (Carrasco-Labra et al 2016, Langendam et al 2016, Santesso et al 2016). They are available through GRADE’s official software package developed to support the GRADE approach: GRADEpro GDT (www.gradepro.org).
Standard Cochrane ‘Summary of findings’ tables include the following elements using one of the accepted formats. Further guidance on each of these is provided in Section 14.1.6.
Ideally, ‘Summary of findings’ tables are supported by more detailed tables (known as ‘evidence profiles’) to which the review may be linked, which provide more detailed explanations. Evidence profiles include the same important health outcomes, and provide greater detail than ‘Summary of findings’ tables of both of the individual considerations feeding into the grading of certainty and of the results of the studies (Guyatt et al 2011a). They ensure that a structured approach is used to rating the certainty of evidence. Although they are rarely published in Cochrane Reviews, evidence profiles are often used, for example, by guideline developers in considering the certainty of the evidence to support guideline recommendations. Review authors will find it easier to develop the ‘Summary of findings’ table by completing the rating of the certainty of evidence in the evidence profile first in GRADEpro GDT. They can then automatically convert this to one of the ‘Summary of findings’ formats in GRADEpro GDT, including an interactive ‘Summary of findings’ for publication.
As a measure of the magnitude of effect for dichotomous outcomes, the ‘Summary of findings’ table should provide a relative measure of effect (e.g. risk ratio, odds ratio, hazard) and measures of absolute risk. For other types of data, an absolute measure alone (such as a difference in means for continuous data) might be sufficient. It is important that the magnitude of effect is presented in a meaningful way, which may require some transformation of the result of a meta-analysis (see also Chapter 15, Section 15.4 and Section 15.5). Reviews with more than one main comparison should include a separate ‘Summary of findings’ table for each comparison.
Figure 14.1.a provides an example of a ‘Summary of findings’ table. Figure 15.1.b provides an alternative format that may further facilitate users’ understanding and interpretation of the review’s findings. Evidence evaluating different formats suggests that the ‘Summary of findings’ table should include a risk difference as a measure of the absolute effect and authors should preferably use a format that includes a risk difference .
Figure 14.1.a Example of a ‘Summary of findings’ table
Summary of findings (for interactive version click here)
Compression stockings compared with no compression stockings for people taking long flights
Patients or population: anyone taking a long flight (lasting more than 6 hours)
Settings: international air travel
Intervention: compression stockings a
Comparison: without stockings
Illustrative comparative risks* (95% CI)
Relative effect (95% CI)
Number of participants (studies)
Certainty of the evidence (GRADE)
Without stockings
With stockings
Symptomatic deep vein thrombosis (DVT)
0 participants developed symptomatic DVT in these studies
Symptomless DVT
Low risk population b
10 per 1000
High risk population b
20 per 1000
Superficial vein thrombosis
13 per 1000
Moderate c
Post-flight values measured on a scale from 0, no oedema, to 10, maximum oedema
The mean oedema score ranged across control groups from
The mean oedema score in the intervention groups was on average
Pulmonary embolus
0 participants developed pulmonary embolus in these studies e
0 participants died in these studies
Adverse effects
The tolerability of the stockings was described as very good with no complaints of side effects in 4 studies f
*The basis for the assumed risk is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the intervention group and the relative effect of the intervention (and its 95% CI).
CI: confidence interval; RR: risk ratio; GRADE: GRADE Working Group grades of evidence (see explanations).
a All the stockings in the nine studies included in this review were below-knee compression stockings. In four studies the compression strength was 20 mmHg to 30 mmHg at the ankle. It was 10 mmHg to 20 mmHg in the other four studies. Stockings come in different sizes. If a stocking is too tight around the knee it can prevent essential venous return causing the blood to pool around the knee. Compression stockings should be fitted properly. A stocking that is too tight could cut into the skin on a long flight and potentially cause ulceration and increased risk of DVT. Some stockings can be slightly thicker than normal leg covering and can be potentially restrictive with tight foot wear. It is a good idea to wear stockings around the house prior to travel to ensure a good, comfortable fit. Participants put their stockings on two to three hours before the flight in most of the studies. The availability and cost of stockings can vary.
b Two studies recruited high risk participants defined as those with previous episodes of DVT, coagulation disorders, severe obesity, limited mobility due to bone or joint problems, neoplastic disease within the previous two years, large varicose veins or, in one of the studies, participants taller than 190 cm and heavier than 90 kg. The incidence for the seven studies that excluded high risk participants was 1.45% and the incidence for the two studies that recruited high-risk participants (with at least one risk factor) was 2.43%. We have used 10 and 30 per 1000 to express different risk strata, respectively.
c The confidence interval crosses no difference and does not rule out a small increase.
d The measurement of oedema was not validated (indirectness of the outcome) or blinded to the intervention (risk of bias).
e If there are very few or no events and the number of participants is large, judgement about the certainty of evidence (particularly judgements about imprecision) may be based on the absolute effect. Here the certainty rating may be considered ‘high’ if the outcome was appropriately assessed and the event, in fact, did not occur in 2821 studied participants.
f None of the other studies reported adverse effects, apart from four cases of superficial vein thrombosis in varicose veins in the knee region that were compressed by the upper edge of the stocking in one study.
Figure 14.1.b Example of alternative ‘Summary of findings’ table
Summary of findings (for interactive version click here):
Probiotics compared to no probiotics as an adjunct to antibiotics in children
Patient or population: children given antibiotics
Settings: inpatients and outpatient
Intervention: probiotics
Comparison: no probiotics
No of participants (studies)
Relative effects
(95% CI)
Anticipated absolute effects* (95% CI)
Certainty of the evidence
(GRADE)
Without probiotics
With probiotics
Incidence of diarrhoea: Probiotic dose 5 billion CFU/day
Follow-up: 10 days to 3 months
moderate b
Due to risk of bias
Probably decreases the incidence of diarrhoea.
13.4% fewer children a
(10.1 to 15.8 fewer)
Children > 5 years
Children > 5 years
Due to risk of bias and imprecision
May decrease the incidence of diarrhoea.
2.2% fewer children a
(5.3 fewer to 2.4 more)
Adverse events d
Follow-up: 10 to 44 days
1575 (11 studies)
0.5% more adverse events e
(1 fewer to 2 more)
Due to risk of bias and inconsistency
There may be little or no difference in adverse events.
Duration of diarrhoea
Follow-up: 10 days to 3 months
The mean duration of diarrhoea without probiotics was 4 days.
0.6 fewer days
(1.18 to 0.02 fewer days)
Due to imprecision and inconsistency
May decrease the duration of diarrhoea.
Stools per day
Follow-up: 10 days to 3 months
The mean stools per day without probiotics was 2.5 stools per day.
0.3 fewer stools per day
Due to imprecision and inconsistency
There may be little or no difference in stools per day.
*The basis for the risk in the control group (e.g. the median control group risk across studies) is provided in footnotes. The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; RR: risk ratio.
EXPLANATIONS
a Control group risk estimates come from pooled estimates of control groups. Relative effect based on available case analysis
b High risk of bias due to high loss to follow-up.
c Imprecision due to few events and confidence intervals include appreciable benefit or harm.
d Side effects: rash, nausea, flatulence, vomiting, increased phlegm, chest pain, constipation, taste disturbance and low appetite.
e Risks were calculated from pooled risk differences.
f High risk of bias. Only 11 of 16 trials reported on adverse events, suggesting a selective reporting bias.
g Serious inconsistency. Numerous probiotic agents and doses were evaluated amongst a relatively small number of trials, limiting our ability to draw conclusions on the safety of the many probiotics agents and doses administered.
h Serious unexplained inconsistency (large heterogeneity I 2 = 79%, P value [P = 0.04], point estimates and confidence intervals vary considerably).
i Serious imprecision. The upper bound of 0.02 fewer days of diarrhoea is not considered patient important.
j Serious unexplained inconsistency (large heterogeneity I 2 = 78%, P value [P = 0.05], point estimates and confidence intervals vary considerably).
k Serious imprecision. The 95% confidence interval includes no effect and lower bound of 0.60 stools per day is of questionable patient importance.
The GRADE Working Group’s software, GRADEpro GDT (www.gradepro.org), including GRADE’s interactive handbook, is available to assist review authors in the preparation of ‘Summary of findings’ tables. GRADEpro can use data on the comparator group risk and the effect estimate (entered by the review authors or imported from files generated in RevMan) to produce the relative effects and absolute risks associated with experimental interventions. In addition, it leads the user through the process of a GRADE assessment, and produces a table that can be used as a standalone table in a review (including by direct import into software such as RevMan or integration with RevMan Web), or an interactive ‘Summary of findings’ table (see help resources in GRADEpro).
‘Summary of findings’ tables should include both absolute and relative measures of effect for dichotomous outcomes. Risk ratios, odds ratios and risk differences are different ways of comparing two groups with dichotomous outcome data (see Chapter 6, Section 6.4.1). Furthermore, there are two distinct risk ratios, depending on which event (e.g. ‘yes’ or ‘no’) is the focus of the analysis (see Chapter 6, Section 6.4.1.5). In the presence of a non-zero intervention effect, any variation across studies in the comparator group risks (i.e. variation in the risk of the event occurring without the intervention of interest, for example in different populations) makes it impossible for more than one of these measures to be truly the same in every study.
It has long been assumed in epidemiology that relative measures of effect are more consistent than absolute measures of effect from one scenario to another. There is empirical evidence to support this assumption (Engels et al 2000, Deeks and Altman 2001, Furukawa et al 2002). For this reason, meta-analyses should generally use either a risk ratio or an odds ratio as a measure of effect (see Chapter 10, Section 10.4.3). Correspondingly, a single estimate of relative effect is likely to be a more appropriate summary than a single estimate of absolute effect. If a relative effect is indeed consistent across studies, then different comparator group risks will have different implications for absolute benefit. For instance, if the risk ratio is consistently 0.75, then the experimental intervention would reduce a comparator group risk of 80% to 60% in the intervention group (an absolute risk reduction of 20 percentage points), but would also reduce a comparator group risk of 20% to 15% in the intervention group (an absolute risk reduction of 5 percentage points).
‘Summary of findings’ tables are built around the assumption of a consistent relative effect. It is therefore important to consider the implications of this effect for different comparator group risks (these can be derived or estimated from a number of sources, see Section 14.1.6.3), which may require an assessment of the certainty of evidence for prognostic evidence (Spencer et al 2012, Iorio et al 2015). For any comparator group risk, it is possible to estimate a corresponding intervention group risk (i.e. the absolute risk with the intervention) from the meta-analytic risk ratio or odds ratio. Note that the numbers provided in the ‘Corresponding risk’ column are specific to the ‘risks’ in the adjacent column.
For the meta-analytic risk ratio (RR) and assumed comparator risk (ACR) the corresponding intervention risk is obtained as:
.
As an example, in Figure 14.1.a, the meta-analytic risk ratio for symptomless deep vein thrombosis (DVT) is RR = 0.10 (95% CI 0.04 to 0.26). Assuming a comparator risk of ACR = 10 per 1000 = 0.01, we obtain:
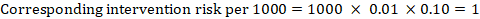
.
For the meta-analytic odds ratio (OR) and assumed comparator risk, ACR, the corresponding intervention risk is obtained as:
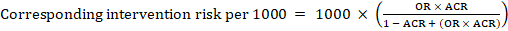
.
Upper and lower confidence limits for the corresponding intervention risk are obtained by replacing RR or OR by their upper and lower confidence limits, respectively (e.g. replacing 0.10 with 0.04, then with 0.26, in the example). Such confidence intervals do not incorporate uncertainty in the assumed comparator risks.
When dealing with risk ratios, it is critical that the same definition of ‘event’ is used as was used for the meta-analysis. For example, if the meta-analysis focused on ‘death’ (as opposed to survival) as the event, then corresponding risks in the ‘Summary of findings’ table must also refer to ‘death’.
In (rare) circumstances in which there is clear rationale to assume a consistent risk difference in the meta-analysis, in principle it is possible to present this for relevant ‘assumed risks’ and their corresponding risks, and to present the corresponding (different) relative effects for each assumed risk.
The risk difference expresses the difference between the ACR and the corresponding intervention risk (or the difference between the experimental and the comparator intervention).
For the meta-analytic risk ratio (RR) and assumed comparator risk (ACR) the corresponding risk difference is obtained as (note that risks can also be expressed using percentage or percentage points):
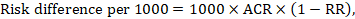
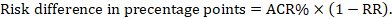
As an example, in Figure 14.1.b the meta-analytic risk ratio is 0.41 (95% CI 0.29 to 0.55) for diarrhoea in children less than 5 years of age. Assuming a comparator group risk of 22.3% we obtain:
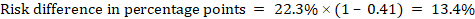
.
For the meta-analytic odds ratio (OR) and assumed comparator risk (ACR) the absolute risk difference is obtained as (percentage points):
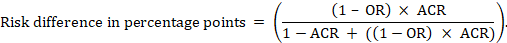
Upper and lower confidence limits for the absolute risk difference are obtained by re-running the calculation above while replacing RR or OR by their upper and lower confidence limits, respectively (e.g. replacing 0.41 with 0.28, then with 0.55, in the example). Such confidence intervals do not incorporate uncertainty in the assumed comparator risks.
Time-to-event outcomes measure whether and when a particular event (e.g. death) occurs (van Dalen et al 2007). The impact of the experimental intervention relative to the comparison group on time-to-event outcomes is usually measured using a hazard ratio (HR) (see Chapter 6, Section 6.8.1).
A hazard ratio expresses a relative effect estimate. It may be used in various ways to obtain absolute risks and other interpretable quantities for a specific population. Here we describe how to re-express hazard ratios in terms of: (i) absolute risk of event-free survival within a particular period of time; (ii) absolute risk of an event within a particular period of time; and (iii) median time to the event. All methods are built on an assumption of consistent relative effects (i.e. that the hazard ratio does not vary over time).
(i) Absolute risk of event-free survival within a particular period of time Event-free survival (e.g. overall survival) is commonly reported by individual studies. To obtain absolute effects for time-to-event outcomes measured as event-free survival, the summary HR can be used in conjunction with an assumed proportion of patients who are event-free in the comparator group (Tierney et al 2007). This proportion of patients will be specific to a period of time of observation. However, it is not strictly necessary to specify this period of time. For instance, a proportion of 50% of event-free patients might apply to patients with a high event rate observed over 1 year, or to patients with a low event rate observed over 2 years.
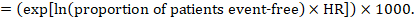
As an example, suppose the meta-analytic hazard ratio is 0.42 (95% CI 0.25 to 0.72). Assuming a comparator group risk of event-free survival (e.g. for overall survival people being alive) at 2 years of ACR = 900 per 1000 = 0.9 we obtain:
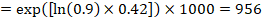
so that that 956 per 1000 people will be alive with the experimental intervention at 2 years. The derivation of the risk should be explained in a comment or footnote.
(ii) Absolute risk of an event within a particular period of time To obtain this absolute effect, again the summary HR can be used (Tierney et al 2007):
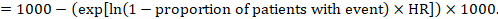
In the example, suppose we assume a comparator group risk of events (e.g. for mortality, people being dead) at 2 years of ACR = 100 per 1000 = 0.1. We obtain:
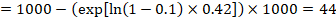
so that that 44 per 1000 people will be dead with the experimental intervention at 2 years.
(iii) Median time to the event Instead of absolute numbers, the time to the event in the intervention and comparison groups can be expressed as median survival time in months or years. To obtain median survival time the pooled HR can be applied to an assumed median survival time in the comparator group (Tierney et al 2007):
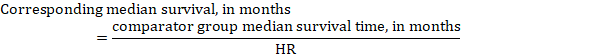
In the example, assuming a comparator group median survival time of 80 months, we obtain:
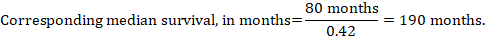
For all three of these options for re-expressing results of time-to-event analyses, upper and lower confidence limits for the corresponding intervention risk are obtained by replacing HR by its upper and lower confidence limits, respectively (e.g. replacing 0.42 with 0.25, then with 0.72, in the example). Again, as for dichotomous outcomes, such confidence intervals do not incorporate uncertainty in the assumed comparator group risks. This is of special concern for long-term survival with a low or moderate mortality rate and a corresponding high number of censored patients (i.e. a low number of patients under risk and a high censoring rate).
The title of each ‘Summary of findings’ table should specify the healthcare question, framed in terms of the population and making it clear exactly what comparison of interventions are made. In Figure 14.1.a, the population is people taking long aeroplane flights, the intervention is compression stockings, and the control is no compression stockings.
The first rows of each ‘Summary of findings’ table should provide the following ‘header’ information:
Patients or population This further clarifies the population (and possibly the subpopulations) of interest and ideally the magnitude of risk of the most crucial adverse outcome at which an intervention is directed. For instance, people on a long-haul flight may be at different risks for DVT; those using selective serotonin reuptake inhibitors (SSRIs) might be at different risk for side effects; while those with atrial fibrillation may be at low ( < 1%), moderate (1% to 4%) or high (>4%) yearly risk of stroke.
Setting This should state any specific characteristics of the settings of the healthcare question that might limit the applicability of the summary of findings to other settings (e.g. primary care in Europe and North America).
Intervention The experimental intervention.
Comparison The comparator intervention (including no specific intervention).
The rows of a ‘Summary of findings’ table should include all desirable and undesirable health outcomes (listed in order of importance) that are essential for decision making, up to a maximum of seven outcomes. If there are more outcomes in the review, review authors will need to omit the less important outcomes from the table, and the decision selecting which outcomes are critical or important to the review should be made during protocol development (see Chapter 3). Review authors should provide time frames for the measurement of the outcomes (e.g. 90 days or 12 months) and the type of instrument scores (e.g. ranging from 0 to 100).
Note that review authors should include the pre-specified critical and important outcomes in the table whether data are available or not. However, they should be alert to the possibility that the importance of an outcome (e.g. a serious adverse effect) may only become known after the protocol was written or the analysis was carried out, and should take appropriate actions to include these in the ‘Summary of findings’ table.
The ‘Summary of findings’ table can include effects in subgroups of the population for different comparator risks and effect sizes separately. For instance, in Figure 14.1.b effects are presented for children younger and older than 5 years separately. Review authors may also opt to produce separate ‘Summary of findings’ tables for different populations.
Review authors should include serious adverse events, but it might be possible to combine minor adverse events as a single outcome, and describe this in an explanatory footnote (note that it is not appropriate to add events together unless they are independent, that is, a participant who has experienced one adverse event has an unaffected chance of experiencing the other adverse event).
Outcomes measured at multiple time points represent a particular problem. In general, to keep the table simple, review authors should present multiple time points only for outcomes critical to decision making, where either the result or the decision made are likely to vary over time. The remainder should be presented at a common time point where possible.
Review authors can present continuous outcome measures in the ‘Summary of findings’ table and should endeavour to make these interpretable to the target audience. This requires that the units are clear and readily interpretable, for example, days of pain, or frequency of headache, and the name and scale of any measurement tools used should be stated (e.g. a Visual Analogue Scale, ranging from 0 to 100). However, many measurement instruments are not readily interpretable by non-specialist clinicians or patients, for example, points on a Beck Depression Inventory or quality of life score. For these, a more interpretable presentation might involve converting a continuous to a dichotomous outcome, such as >50% improvement (see Chapter 15, Section 15.5).
Review authors should provide up to three typical risks for participants receiving the comparator intervention. For dichotomous outcomes, we recommend that these be presented in the form of the number of people experiencing the event per 100 or 1000 people (natural frequency) depending on the frequency of the outcome. For continuous outcomes, this would be stated as a mean or median value of the outcome measured.
Estimated or assumed comparator intervention risks could be based on assessments of typical risks in different patient groups derived from the review itself, individual representative studies in the review, or risks derived from a systematic review of prognosis studies or other sources of evidence which may in turn require an assessment of the certainty for the prognostic evidence (Spencer et al 2012, Iorio et al 2015). Ideally, risks would reflect groups that clinicians can easily identify on the basis of their presenting features.
An explanatory footnote should specify the source or rationale for each comparator group risk, including the time period to which it corresponds where appropriate. In Figure 14.1.a, clinicians can easily differentiate individuals with risk factors for deep venous thrombosis from those without. If there is known to be little variation in baseline risk then review authors may use the median comparator group risk across studies. If typical risks are not known, an option is to choose the risk from the included studies, providing the second highest for a high and the second lowest for a low risk population.
For dichotomous outcomes, review authors should provide a corresponding absolute risk for each comparator group risk, along with a confidence interval. This absolute risk with the (experimental) intervention will usually be derived from the meta-analysis result presented in the relative effect column (see Section 14.1.6.6). Formulae are provided in Section 14.1.5. Review authors should present the absolute effect in the same format as the risks with comparator intervention (see Section 14.1.6.3), for example as the number of people experiencing the event per 1000 people.
For continuous outcomes, a difference in means or standardized difference in means should be presented with its confidence interval. These will typically be obtained directly from a meta-analysis. Explanatory text should be used to clarify the meaning, as in Figures 14.1.a and 14.1.b.
For dichotomous outcomes, the risk difference can be provided using one of the ‘Summary of findings’ table formats as an additional option (see Figure 14.1.b). This risk difference expresses the difference between the experimental and comparator intervention and will usually be derived from the meta-analysis result presented in the relative effect column (see Section 14.1.6.6). Formulae are provided in Section 14.1.5. Review authors should present the risk difference in the same format as assumed and corresponding risks with comparator intervention (see Section 14.1.6.3); for example, as the number of people experiencing the event per 1000 people or as percentage points if the assumed and corresponding risks are expressed in percentage.
For continuous outcomes, if the ‘Summary of findings’ table includes this option, the mean difference can be presented here and the ‘corresponding risk’ column left blank (see Figure 14.1.b).
The relative effect will typically be a risk ratio or odds ratio (or occasionally a hazard ratio) with its accompanying 95% confidence interval, obtained from a meta-analysis performed on the basis of the same effect measure. Risk ratios and odds ratios are similar when the comparator intervention risks are low and effects are small, but may differ considerably when comparator group risks increase. The meta-analysis may involve an assumption of either fixed or random effects, depending on what the review authors consider appropriate, and implying that the relative effect is either an estimate of the effect of the intervention, or an estimate of the average effect of the intervention across studies, respectively.
This column should include the number of participants assessed in the included studies for each outcome and the corresponding number of studies that contributed these participants.
Review authors should comment on the certainty of the evidence (also known as quality of the body of evidence or confidence in the effect estimates). Review authors should use the specific evidence grading system developed by the GRADE Working Group (Atkins et al 2004, Guyatt et al 2008, Guyatt et al 2011a), which is described in detail in Section 14.2. The GRADE approach categorizes the certainty in a body of evidence as ‘high’, ‘moderate’, ‘low’ or ‘very low’ by outcome. This is a result of judgement, but the judgement process operates within a transparent structure. As an example, the certainty would be ‘high’ if the summary were of several randomized trials with low risk of bias, but the rating of certainty becomes lower if there are concerns about risk of bias, inconsistency, indirectness, imprecision or publication bias. Judgements other than of ‘high’ certainty should be made transparent using explanatory footnotes or the ‘Comments’ column in the ‘Summary of findings’ table (see Section 14.1.6.10).
The aim of the ‘Comments’ field is to help interpret the information or data identified in the row. For example, this may be on the validity of the outcome measure or the presence of variables that are associated with the magnitude of effect. Important caveats about the results should be flagged here. Not all rows will need comments, and it is best to leave a blank if there is nothing warranting a comment.
Detailed explanations should be included as footnotes to support the judgements in the ‘Summary of findings’ table, such as the overall GRADE assessment. The explanations should describe the rationale for important aspects of the content. Table 14.1.a lists guidance for useful explanations. Explanations should be concise, informative, relevant, easy to understand and accurate. If explanations cannot be sufficiently described in footnotes, review authors should provide further details of the issues in the Results and Discussion sections of the review.
Table 14.1.a Guidance for providing useful explanations in ‘Summary of findings’ (SoF) tables. Adapted from Santesso et al (2016)
General guidance
Domain-specific guidance for writing useful explanations
The Grades of Recommendation, Assessment, Development and Evaluation Working Group (GRADE Working Group) has developed a system for grading the certainty of evidence (Schünemann et al 2003, Atkins et al 2004, Schünemann et al 2006, Guyatt et al 2008, Guyatt et al 2011a). Over 100 organizations including the World Health Organization (WHO), the American College of Physicians, the American Society of Hematology (ASH), the Canadian Agency for Drugs and Technology in Health (CADTH) and the National Institutes of Health and Clinical Excellence (NICE) in the UK have adopted the GRADE system (www.gradeworkinggroup.org).
Cochrane has also formally adopted this approach, and all Cochrane Reviews should use GRADE to evaluate the certainty of evidence for important outcomes (see MECIR Box 14.2.a).
MECIR Box 14.2.a Relevant expectations for conduct of intervention reviews
C74: Assessing the certainty of the body of evidence (Mandatory)
Use the five GRADE considerations (risk of bias, consistency of effect, imprecision, indirectness and publication bias) to assess the certainty of the body of evidence for each outcome, and to draw conclusions about the certainty of evidence within the text of the review.
GRADE is the most widely used approach for summarizing confidence in effects of interventions by outcome across studies. It is preferable to use the online GRADEpro tool, and to use it as described in the help system of the software. This should help to ensure that author teams are accessing the same information to inform their judgements. Ideally, two people working independently should assess the certainty of the body of evidence and reach a consensus view on any downgrading decisions. The five GRADE considerations should be addressed irrespective of whether the review includes a ‘Summary of findings’ table. It is helpful to draw on this information in the Discussion, in the Authors’ conclusions and to convey the certainty in the evidence in the Abstract and Plain language summary.
C75: Justifying assessments of the certainty of the body of evidence (Mandatory)
Justify and document all assessments of the certainty of the body of evidence (e.g. downgrading or upgrading using GRADE).
The adoption of a structured approach ensures transparency in formulating an interpretation of the evidence, and the result is more informative to the user.
For systematic reviews, the GRADE approach defines the certainty of a body of evidence as the extent to which one can be confident that an estimate of effect or association is close to the quantity of specific interest. Assessing the certainty of a body of evidence involves consideration of within- and across-study risk of bias (limitations in study design and execution or methodological quality), inconsistency (or heterogeneity), indirectness of evidence, imprecision of the effect estimates and risk of publication bias (see Section 14.2.2), as well as domains that may increase our confidence in the effect estimate (as described in Section 14.2.3). The GRADE system entails an assessment of the certainty of a body of evidence for each individual outcome. Judgements about the domains that determine the certainty of evidence should be described in the results or discussion section and as part of the ‘Summary of findings’ table.
The GRADE approach specifies four levels of certainty (Figure 14.2.a). For interventions, including diagnostic and other tests that are evaluated as interventions (Schünemann et al 2008b, Schünemann et al 2008a, Balshem et al 2011, Schünemann et al 2012), the starting point for rating the certainty of evidence is categorized into two types:
There are many instances in which review authors rely on information from NRSI, in particular to evaluate potential harms (see Chapter 24). In addition, review authors can obtain relevant data from both randomized trials and NRSI, with each type of evidence complementing the other (Schünemann et al 2013).
In GRADE, a body of evidence from randomized trials begins with a high-certainty rating while a body of evidence from NRSI begins with a low-certainty rating. The lower rating with NRSI is the result of the potential bias induced by the lack of randomization (i.e. confounding and selection bias).
However, when using the new Risk Of Bias In Non-randomized Studies of Interventions (ROBINS-I) tool (Sterne et al 2016), an assessment tool that covers the risk of bias due to lack of randomization, all studies may start as high certainty of the evidence (Schünemann et al 2018). The approach of starting all study designs (including NRSI) as high certainty does not conflict with the initial GRADE approach of starting the rating of NRSI as low certainty evidence. This is because a body of evidence from NRSI should generally be downgraded by two levels due to the inherent risk of bias associated with the lack of randomization, namely confounding and selection bias. Not downgrading NRSI from high to low certainty needs transparent and detailed justification for what mitigates concerns about confounding and selection bias (Schünemann et al 2018). Very few examples of where not rating down by two levels is appropriate currently exist.
The highest certainty rating is a body of evidence when there are no concerns in any of the GRADE factors listed in Figure 14.2.a. Review authors often downgrade evidence to moderate, low or even very low certainty evidence, depending on the presence of the five factors in Figure 14.2.a. Usually, certainty rating will fall by one level for each factor, up to a maximum of three levels for all factors. If there are very severe problems for any one domain (e.g. when assessing risk of bias, all studies were unconcealed, unblinded and lost over 50% of their patients to follow-up), evidence may fall by two levels due to that factor alone. It is not possible to rate lower than ‘very low certainty’ evidence.
Review authors will generally grade evidence from sound non-randomized studies as low certainty, even if ROBINS-I is used. If, however, such studies yield large effects and there is no obvious bias explaining those effects, review authors may rate the evidence as moderate or – if the effect is large enough – even as high certainty (Figure 14.2.a). The very low certainty level is appropriate for, but is not limited to, studies with critical problems and unsystematic clinical observations (e.g. case series or case reports).
Figure 14.2.a Levels of the certainty of a body of evidence in the GRADE approach. *Upgrading criteria are usually applicable to non-randomized studies only (but exceptions exist).
1.
Establish initial
level of certainty
2.
Consider lowering or raising
level of certainty
3.
Final level of
certainty rating
Study design
Initial certainty
in an estimate of effect
Reasons for considering lowering
or raising certainty
Certainty
in an estimate of effect
across those considerations
Randomized trials or studies evaluated with ROBINS-I
High
certainty
Risk of bias
Inconsistency
Indirectness
Imprecision
Publication bias
Large effect
Dose response
All plausible
confounding and bias:
Observational studies not using ROBINS-I
Low
certainty
We now describe in more detail the five reasons (or domains) for downgrading the certainty of a body of evidence for a specific outcome. In each case, if no reason is found for downgrading the evidence, it should be classified as 'no limitation or not serious' (not important enough to warrant downgrading). If a reason is found for downgrading the evidence, it should be classified as 'serious' (downgrading the certainty rating by one level) or 'very serious' (downgrading the certainty grade by two levels). For non-randomized studies assessed with ROBINS-I, rating down by three levels should be classified as 'extremely' serious.
(1) Risk of bias or limitations in the detailed design and implementation
Our confidence in an estimate of effect decreases if studies suffer from major limitations that are likely to result in a biased assessment of the intervention effect. For randomized trials, these methodological limitations include failure to generate a random sequence, lack of allocation sequence concealment, lack of blinding (particularly with subjective outcomes that are highly susceptible to biased assessment), a large loss to follow-up or selective reporting of outcomes. Chapter 8 provides a discussion of study-level assessments of risk of bias in the context of a Cochrane Review, and proposes an approach to assessing the risk of bias for an outcome across studies as ‘Low’ risk of bias, ‘Some concerns’ and ‘High’ risk of bias for randomized trials. Levels of ‘Low’. ‘Moderate’, ‘Serious’ and ‘Critical’ risk of bias arise for non-randomized studies assessed with ROBINS-I (Chapter 25). These assessments should feed directly into this GRADE domain. In particular, ‘Low’ risk of bias would indicate ‘no limitation’; ‘Some concerns’ would indicate either ‘no limitation’ or ‘serious limitation’; and ‘High’ risk of bias would indicate either ‘serious limitation’ or ‘very serious limitation’. ‘Critical’ risk of bias on ROBINS-I would indicate extremely serious limitations in GRADE. Review authors should use their judgement to decide between alternative categories, depending on the likely magnitude of the potential biases.
Every study addressing a particular outcome will differ, to some degree, in the risk of bias. Review authors should make an overall judgement on whether the certainty of evidence for an outcome warrants downgrading on the basis of study limitations. The assessment of study limitations should apply to the studies contributing to the results in the ‘Summary of findings’ table, rather than to all studies that could potentially be included in the analysis. We have argued in Chapter 7, Section 7.6.2, that the primary analysis should be restricted to studies at low (or low and unclear) risk of bias where possible.
Table 14.2.a presents the judgements that must be made in going from assessments of the risk of bias to judgements about study limitations for each outcome included in a ‘Summary of findings’ table. A rating of high certainty evidence can be achieved only when most evidence comes from studies that met the criteria for low risk of bias. For example, of the 22 studies addressing the impact of beta-blockers on mortality in patients with heart failure, most probably or certainly used concealed allocation of the sequence, all blinded at least some key groups and follow-up of randomized patients was almost complete (Brophy et al 2001). The certainty of evidence might be downgraded by one level when most of the evidence comes from individual studies either with a crucial limitation for one item, or with some limitations for multiple items. An example of very serious limitations, warranting downgrading by two levels, is provided by evidence on surgery versus conservative treatment in the management of patients with lumbar disc prolapse (Gibson and Waddell 2007). We are uncertain of the benefit of surgery in reducing symptoms after one year or longer, because the one study included in the analysis had inadequate concealment of the allocation sequence and the outcome was assessed using a crude rating by the surgeon without blinding.
(2) Unexplained heterogeneity or inconsistency of results
When studies yield widely differing estimates of effect (heterogeneity or variability in results), investigators should look for robust explanations for that heterogeneity. For instance, drugs may have larger relative effects in sicker populations or when given in larger doses. A detailed discussion of heterogeneity and its investigation is provided in Chapter 10, Section 10.10 and Section 10.11. If an important modifier exists, with good evidence that important outcomes are different in different subgroups (which would ideally be pre-specified), then a separate ‘Summary of findings’ table may be considered for a separate population. For instance, a separate ‘Summary of findings’ table would be used for carotid endarterectomy in symptomatic patients with high grade stenosis (70% to 99%) in which the intervention is, in the hands of the right surgeons, beneficial, and another (if review authors considered it relevant) for asymptomatic patients with low grade stenosis (less than 30%) in which surgery appears harmful (Orrapin and Rerkasem 2017). When heterogeneity exists and affects the interpretation of results, but review authors are unable to identify a plausible explanation with the data available, the certainty of the evidence decreases.
(3) Indirectness of evidence
Two types of indirectness are relevant. First, a review comparing the effectiveness of alternative interventions (say A and B) may find that randomized trials are available, but they have compared A with placebo and B with placebo. Thus, the evidence is restricted to indirect comparisons between A and B. Where indirect comparisons are undertaken within a network meta-analysis context, GRADE for network meta-analysis should be used (see Chapter 11, Section 11.5).
Second, a review may find randomized trials that meet eligibility criteria but address a restricted version of the main review question in terms of population, intervention, comparator or outcomes. For example, suppose that in a review addressing an intervention for secondary prevention of coronary heart disease, most identified studies happened to be in people who also had diabetes. Then the evidence may be regarded as indirect in relation to the broader question of interest because the population is primarily related to people with diabetes. The opposite scenario can equally apply: a review addressing the effect of a preventive strategy for coronary heart disease in people with diabetes may consider studies in people without diabetes to provide relevant, albeit indirect, evidence. This would be particularly likely if investigators had conducted few if any randomized trials in the target population (e.g. people with diabetes). Other sources of indirectness may arise from interventions studied (e.g. if in all included studies a technical intervention was implemented by expert, highly trained specialists in specialist centres, then evidence on the effects of the intervention outside these centres may be indirect), comparators used (e.g. if the comparator groups received an intervention that is less effective than standard treatment in most settings) and outcomes assessed (e.g. indirectness due to surrogate outcomes when data on patient-important outcomes are not available, or when investigators seek data on quality of life but only symptoms are reported). Review authors should make judgements transparent when they believe downgrading is justified, based on differences in anticipated effects in the group of primary interest. Review authors may be aided and increase transparency of their judgements about indirectness if they use Table 14.2.b available in the GRADEpro GDT software (Schünemann et al 2013).
(4) Imprecision of results
When studies include few participants or few events, and thus have wide confidence intervals, review authors can lower their rating of the certainty of the evidence. The confidence intervals included in the ‘Summary of findings’ table will provide readers with information that allows them to make, to some extent, their own rating of precision. Review authors can use a calculation of the optimal information size (OIS) or review information size (RIS), similar to sample size calculations, to make judgements about imprecision (Guyatt et al 2011b, Schünemann 2016). The OIS or RIS is calculated on the basis of the number of participants required for an adequately powered individual study. If the 95% confidence interval excludes a risk ratio (RR) of 1.0, and the total number of events or patients exceeds the OIS criterion, precision is adequate. If the 95% CI includes appreciable benefit or harm (an RR of under 0.75 or over 1.25 is often suggested as a very rough guide) downgrading for imprecision may be appropriate even if OIS criteria are met (Guyatt et al 2011b, Schünemann 2016).
(5) High probability of publication bias
The certainty of evidence level may be downgraded if investigators fail to report studies on the basis of results (typically those that show no effect: publication bias) or outcomes (typically those that may be harmful or for which no effect was observed: selective outcome non-reporting bias). Selective reporting of outcomes from among multiple outcomes measured is assessed at the study level as part of the assessment of risk of bias (see Chapter 8, Section 8.7), so for the studies contributing to the outcome in the ‘Summary of findings’ table this is addressed by domain 1 above (limitations in the design and implementation). If a large number of studies included in the review do not contribute to an outcome, or if there is evidence of publication bias, the certainty of the evidence may be downgraded. Chapter 13 provides a detailed discussion of reporting biases, including publication bias, and how it may be tackled in a Cochrane Review. A prototypical situation that may elicit suspicion of publication bias is when published evidence includes a number of small studies, all of which are industry-funded (Bhandari et al 2004). For example, 14 studies of flavanoids in patients with haemorrhoids have shown apparent large benefits, but enrolled a total of only 1432 patients (i.e. each study enrolled relatively few patients) (Alonso-Coello et al 2006). The heavy involvement of sponsors in most of these studies raises questions of whether unpublished studies that suggest no benefit exist (publication bias).
A particular body of evidence can suffer from problems associated with more than one of the five factors listed here, and the greater the problems, the lower the certainty of evidence rating that should result. One could imagine a situation in which randomized trials were available, but all or virtually all of these limitations would be present, and in serious form. A very low certainty of evidence rating would result.
Table 14.2.a Further guidelines for domain 1 (of 5) in a GRADE assessment: going from assessments of risk of bias in studies to judgements about study limitations for main outcomes across studies